Asociación Española de Raquitismos y Osteomalacia Heredados
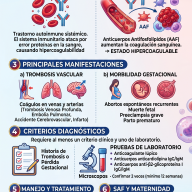
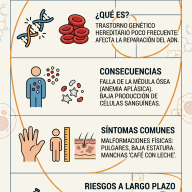
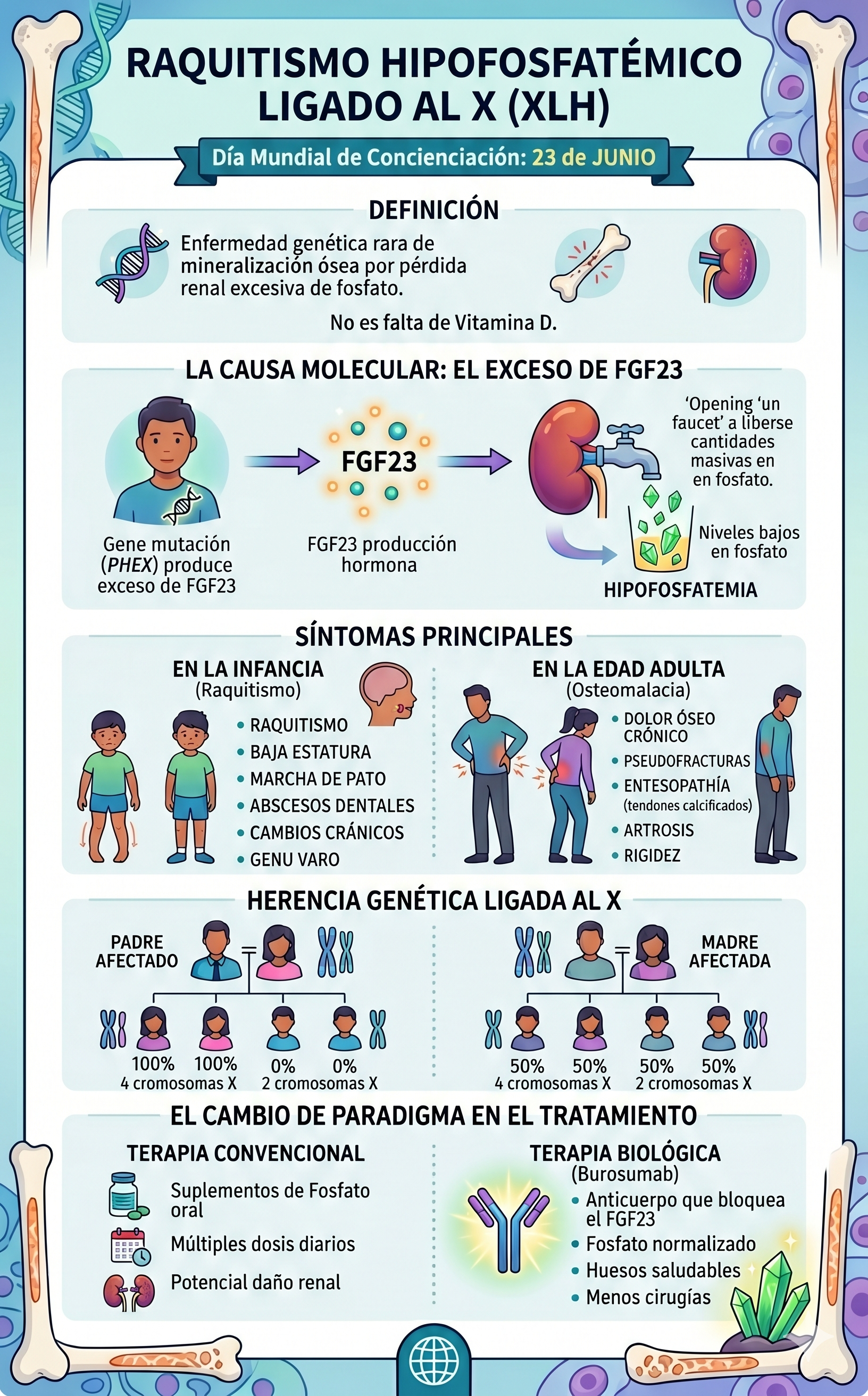
El Raquitismo Hipofosfatémico Ligado al X (XLH) es una enfermedad de origen genético, clasificada dentro del grupo de las enfermedades raras, que afecta de forma directa al desarrollo y la mineralización de los huesos.
A diferencia del raquitismo clásico o común —que la mayoría de las personas asocia con la falta de vitamina D o una desnutrición en la infancia—, el XLH no se puede prevenir ni curar mejorando la dieta o tomando el sol. Su raíz se encuentra en un "error de comunicación" molecular que provoca que el cuerpo elimine de manera masiva un mineral indispensable para el esqueleto: el fosfato.
La causa oculta: El "grifo abierto" de los riñones
Para entender el XLH, debemos imaginar al fósforo (que en el cuerpo circula en forma de fosfato) como los ladrillos que endurecen y dan estructura a nuestros huesos. En condiciones normales, los riñones actúan como un filtro inteligente: retienen el fosfato que el cuerpo necesita y descartan el exceso a través de la orina.
En una persona con XLH, este sistema de filtrado falla debido a una mutación en un gen llamado PHEX, ubicado en el cromosoma X. El proceso biológico se desencadena de la siguiente manera:
- La mutación de PHEX: Al no funcionar correctamente este gen, el cuerpo pierde el control sobre la producción de una hormona llamada FGF23 (Factor de Crecimiento Fibroblástico 23).
- El exceso de FGF23: El organismo empieza a producir esta hormona en cantidades exageradas. La función principal de la FGF23 es ordenarle a los riñones que expulsen el fosfato.
- La pérdida mineral: Al haber demasiada hormona en circulación, los riñones abren sus compuertas por completo y eliminan el fosfato de forma descontrolada (hiperfosfaturia).
Como consecuencia, la concentración de fosfato en la sangre cae a niveles peligrosamente bajos (hipofosfatemia), dejando al esqueleto sin la materia prima necesaria para endurecerse (calcificarse).
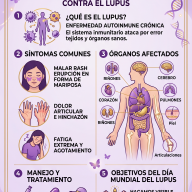
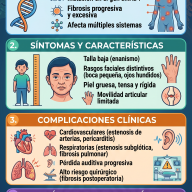
Síntomas y manifestaciones clínicas
La falta de mineralización ósea debilita la estructura ósea general. Dado que los huesos de las piernas deben soportar el peso de todo el cuerpo durante el crecimiento, los impactos físicos más evidentes se concentran en las extremidades inferiores. Sin embargo, la enfermedad afecta al organismo de manera sistémica y evoluciona con la edad.
| Etapa de la vida | Manifestaciones principales |
| Infancia y niñez |
• Genu varo o valgo: Piernas arqueadas notablemente en forma de "O" o en forma de "X". • Retraso en el crecimiento: Talla significativamente baja en comparación con el promedio familiar. • Alteraciones en la marcha: Retraso al empezar a caminar y desarrollo de un andar balanceado (marcha de pato). • Deformidades craneales: Cierre prematuro de las suturas del cráneo (craneosinostosis). |
| Edad adulta |
• Osteomalacia: Ablandamiento severo de los huesos que genera dolor constante y profundo. • Seudofracturas: Pequeñas fisuras en los huesos que no logran consolidar por falta de minerales. • Entesopatía: Calcificación dolorosa de los ligamentos y tendones donde se unen al hueso. • Fatiga y rigidez: Debilidad muscular crónica y artrosis de aparición temprana. |
El impacto en la salud dental: El fosfato también es clave para la formación de la dentina y el esmalte de los dientes. Las personas con XLH suelen sufrir de abscesos (infecciones) dentales espontáneos muy dolorosos, los cuales aparecen en dientes aparentemente sanos y sin caries, debido a la porosidad interna del diente.
El patrón de herencia genética
Al ser una enfermedad ligada al cromosoma X, las probabilidades de transmitir la mutación a la descendencia siguen reglas matemáticas muy específicas dependiendo de cuál de los progenitores esté afectado:
- Si el padre tiene XLH: Al tener un mapa genético XY, el padre transmite su cromosoma Y a sus hijos varones (quienes nacerán sanos) y su único cromosoma X afectado a sus hijas. Por lo tanto, el 100% de sus hijas heredará la enfermedad.
- Si la madre tiene XLH: Al poseer dos cromosomas X (uno sano y uno afectado), la probabilidad de transmitir el cromosoma con la mutación es del 50% en cada embarazo, independientemente de si el bebé es niño o niña.
Nota: En aproximadamente un 20% a 30% de los casos, la enfermedad aparece por primera vez en una familia debido a una mutación espontánea (denominada técnicamente mutación "de novo").
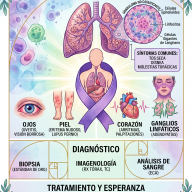
El cambio de paradigma en el tratamiento
Durante décadas, el abordaje médico consistió en una terapia "artesanal" y puramente sintomática: administrar a los pacientes grandes dosis diarias de suplementos de fosfato oral combinados con vitamina D activa (calcitriol). Este tratamiento requería múltiples tomas al día, generaba malestar gastrointestinal y con frecuencia provocaba daños secundarios en los riñones por acumulación de calcio (nefrocalcinosis).
El panorama cambió radicalmente gracias a la medicina de precisión y el desarrollo de la terapia biológica:
- El uso de Burosumab: Es un anticuerpo monoclonal diseñado específicamente para localizar y bloquear la hormona FGF23 sobrante. Al neutralizar este exceso, los riñones recuperan la capacidad de reabsorber el fosfato, normalizando de manera natural sus niveles en la sangre.
Este avance permite que los niños con XLH mineralicen adecuadamente sus huesos durante la etapa crítica del desarrollo, previniendo o corrigiendo en gran medida el arqueamiento de las piernas y reduciendo drásticamente la necesidad de complejas cirugías ortopédicas en el futuro.
últimas noticias sobre Raquitismo Hipofosfatémico Ligado al X
Últimas noticias