Asociación Glut1 y enfermedades que responden a una dieta cetogénica
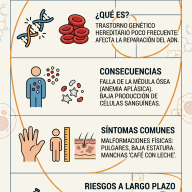
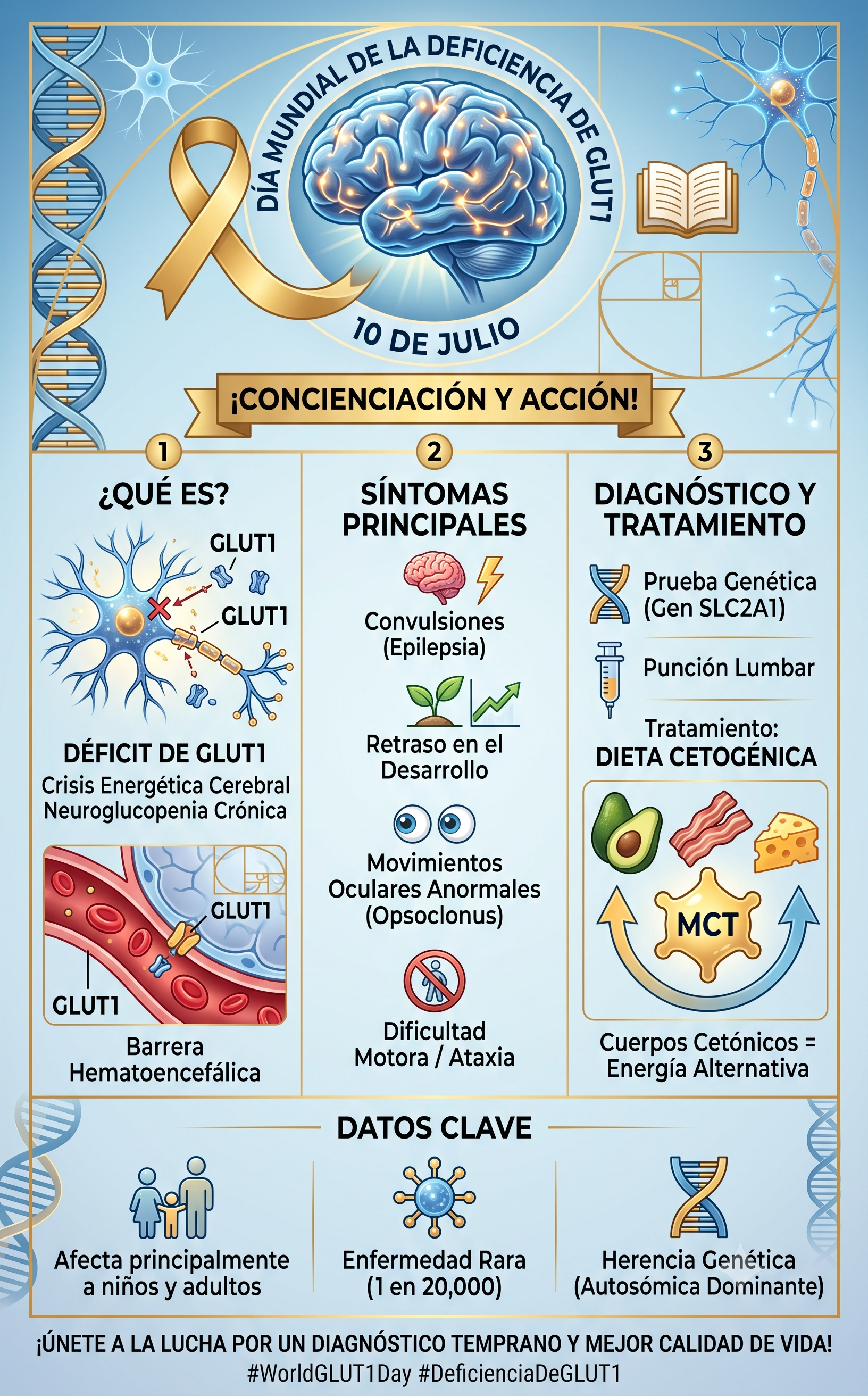
El síndrome de deficiencia del transportador de glucosa tipo 1 (G1D o DSG1) es un trastorno metabólico y neurológico de origen genético, caracterizado por un fallo en el transporte de glucosa a través de la barrera hematoencefálica y las membranas celulares del sistema nervioso central.
Descrito por primera vez por el Dr. Darryl De Vivo en 1991, este síndrome representa un paradigma en la neurología metabólica: una crisis energética cerebral secundaria no a la falta de sustrato en el organismo, sino a la incapacidad estructural para introducirlo en el parénquima cerebral.
1. Fisiopatología y Base Molecular
El cerebro humano adulto representa aproximadamente el 2% del peso corporal, pero consume cerca del 20% de la energía total del organismo, dependiendo casi de forma exclusiva del metabolismo aeróbico de la glucosa. Debido a que la glucosa es una molécula hidrófila, no puede difundirse libremente a través de las membranas lipídicas de la barrera hematoencefálica (BHE). Requiere un mecanismo de difusión facilitada mediada por transportadores específicos.
La proteína GLUT1 (codificada por el gen SLC2A1, ubicado en el cromosoma 1p34.2) es la isoforma del transportador de glucosa predominante en las células endoteliales de los capilares cerebrales y en los astrocitos.
En los individuos con DSG1, la mutación en el gen SLC2A1 (frecuentemente deleciones, mutaciones sin sentido o de cambio de sentido) provoca una pérdida de función de la proteína. Esto reduce drásticamente el flujo de glucosa hacia el espacio intersticial cerebral, cronificando un estado de neuroglucopenia (déficit de glucosa en el tejido cerebral), a pesar de que los niveles de glucemia periférica (en sangre) se mantengan estrictamente normales.
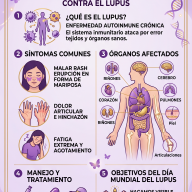
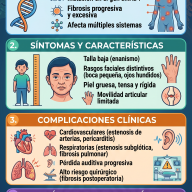
2. Manifestaciones Clínicas y Variabilidad Fenotípica
El espectro clínico del DSG1 es notablemente amplio y evolutivo. Tradicionalmente se clasificaba en fenotipos clásicos y no clásicos, aunque hoy se entiende como un continuo de gravedad.
Fenotipo Clásico
- Crisis Epilépticas Tempranas: Es el signo cardinal en el 80-90% de los casos. Suelen debutar entre el primer y el cuarto mes de vida. Las crisis son polimórficas (ausencias, mioclónicas, tónico-clónicas o focales) y se caracterizan por una marcada resistencia a los fármacos antiepilépticos convencionales.
- Trastornos del Movimiento Complejos: Los pacientes desarrollan una combinación de ataxia (incoordinación motora), distonía (contracciones musculares sostenidas) y coreoatetosis. Estos movimientos anormales suelen presentar una fluctuación diurna y se exacerban con el ayuno, el estrés térmico (fiebre) o el ejercicio.
- Retraso en el Desarrollo Neurocognitivo: Se evidencia microcefalia adquirida (desaceleración del crecimiento cefálico), retraso psicomotor global y disartria o ausencia de lenguaje.
Fenotipo No Clásico (Formas Atenuadas)
Incluye a pacientes que no presentan epilepsia manifiesta, sino predominantemente trastornos del movimiento recurrentes, como la discinesia paroxística inducida por el ejercicio (episodios bruscos de distonía o debilidad desencadenados por el esfuerzo físico) o cuadros que simulan una parálisis cerebral espástica familiar.
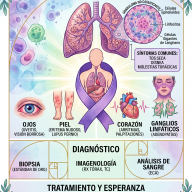
3. Criterios Diagnósticos
El retraso en el diagnóstico del DSG1 es uno de los mayores desafíos clínicos, con una media histórica de varios años desde el debut de los síntomas. El abordaje diagnóstico actual se fundamenta en tres pilares secuenciales:
Análisis del Líquido Cefalorraquídeo (LCR)
La punción lumbar sigue siendo el gold standard bioquímico preliminar. Los hallazgos patognomónicos son:
- Hipoglucorraquia: Concentración de glucosa en el LCR significativamente baja (generalmente inferior a $40\text{ mg/dL}$ o $2.2\text{ mmol/L}$).
- Relación Glucosa LCR/Sangre: Un cociente alterado, típicamente inferior a $0.40$ (en condiciones normales es superior a $0.60$), medido en ayuno.
- Lactato en LCR: Valores normales o disminuidos (habitualmente $< 1.3\text{ mmol/L}$), lo que permite diferenciarlo de las encefalopatías mitocondriales, donde el lactato suele estar elevado.
Confirmación Genética
El secuenciación del gen SLC2A1 identifica mutaciones patogénicas en aproximadamente el 95% de los pacientes con sospecha clínica y bioquímica. La gran mayoría de los casos ocurren por mutaciones de novo (espontáneas), aunque existe un patrón de herencia autosómico dominante bien documentado, y excepcionalmente casos autosómicos recesivos con afectación parcial.
Electroencefalograma (EEG)
Aunque los hallazgos electroencefalográficos son variables, un rasgo altamente orientativo es la presencia de descargas de puntas u ondas lentas generalizadas en estado de ayuno, las cuales muestran una atenuación o normalización inmediata tras la ingesta de alimentos (especialmente carbohidratos).
4. Abordaje Terapéutico: Reprogramación Metabólica
Al tratarse de un defecto estructural del transporte, el tratamiento farmacológico convencional de la epilepsia suele ser ineficaz. El abordaje terapéutico se centra en proporcionar una fuente de energía alternativa que evite la vía del transportador GLUT1.
La Dieta Cetogénica (DC)
Es la terapia médica de elección y debe instaurarse de forma precoz para mitigar el daño neurológico del cerebro en desarrollo.
La DC es un régimen nutricional estricto, calculado en ratios de macronutrientes (típicamente 4:1 o 3:1; es decir, 4 gramos de grasa por cada gramo combinado de proteínas y carbohidratos).
Mecanismo de acción:
Al restringir drásticamente los carbohidratos, el organismo entra en un estado de cetosis controlada. El hígado metaboliza los ácidos grasos transformándolos en cuerpos cetónicos (acetoacetato y beta-hidroxibutirato).
A diferencia de la glucosa, los cuerpos cetónicos atraviesan la barrera hematoencefálica utilizando un sistema de transporte completamente independiente: los transportadores de monocarboxilatos (MCT1 y MCT2). Una vez en el interior de las neuronas y astrocitos, los cuerpos cetónicos ingresan directamente al ciclo de Krebs en las mitocondrias, restaurando la producción de ATP celular y estabilizando las membranas neuronales.
[Ayuno / Dieta Cetogénica] ➔ Hígado ➔ Cuerpos Cetónicos (β-hidroxibutirato) │ (Cruce vía MCT1) ▼ [Barrera Hematoencefálica] │ ▼ Neuronas / Astrocitos │ (Ciclo de Krebs) ▼ Restauración de ATP (Energía)
Sustancias Contraindicadas
Es imperativo que los pacientes con DSG1 eviten fármacos que actúen como inhibidores conocidos del transportador GLUT1, ya que pueden precipitar crisis neuroglucopénicas graves. Entre ellos se encuentran el fenobarbital, el ácido valproico, la cafeína en dosis elevadas y ciertos antagonistas del calcio.
5. Pronóstico y Perspectivas Futuras
El pronóstico del DSG1 está intrínsecamente ligado a la precocidad del diagnóstico y al inicio de la terapia cetogénica. Si se interviene en etapas tempranas de la lactancia, se puede prevenir el desarrollo de discapacidad intelectual grave y lograr un control óptimo de las crisis epilépticas. Sin embargo, los trastornos del movimiento tienden a ser más refractarios a largo plazo y requieren terapias físicas y ocupacionales continuadas.
La investigación biomédica actual se orienta hacia el desarrollo de pequeñas moléculas capaces de modular o aumentar la expresión del transportador GLUT1 residual, así como hacia ensayos clínicos preliminares de terapia génica mediante vectores virales (AAV) diseñados para restaurar la funcionalidad del gen SLC2A1 directamente en el endotelio capilar cerebral.
(Contenido generado con Gemini IA)
últimas noticias sobre Deficiencia de Glut1
Últimas noticias