Asociación Balear de la enfermedad de Andrade | Asociación Valverdeña de la Enfermedad de Andrade
Aquí tienes todo el informe unificado, estructurado y en texto plano enriquecido (Markdown) para que puedas seleccionarlo, copiarlo y pegarlo fácilmente en cualquier documento, nota o correo electrónico sin elementos visuales bloqueantes.
INFORME COMPLETO: ENFERMEDAD DE ANDRADE Y TRATAMIENTOS DE ÚLTIMA GENERACIÓN (ARN de Interferencia)
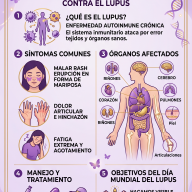
La enfermedad de Andrade, conocida médicamente como Polineuropatía Amiloidótica Familiar (PAF) o amiloidosis hereditaria por transtiretina (AhTTR), es una enfermedad genética, hereditaria y neurodegenerativa poco frecuente. Se calcula que afecta a unas 10.000 personas en todo el mundo, concentrándose de forma muy intensa en focos geográficos específicos.
Fue descrita por primera vez en 1952 por el neurólogo portugués Corino de Andrade en comunidades pesqueras. Hoy en día, la ciencia comprende con precisión su origen molecular y dispone de tratamientos avanzados que han cambiado radicalmente el pronóstico de la enfermedad.
1. El Origen Molecular y Mecanismo de la Enfermedad
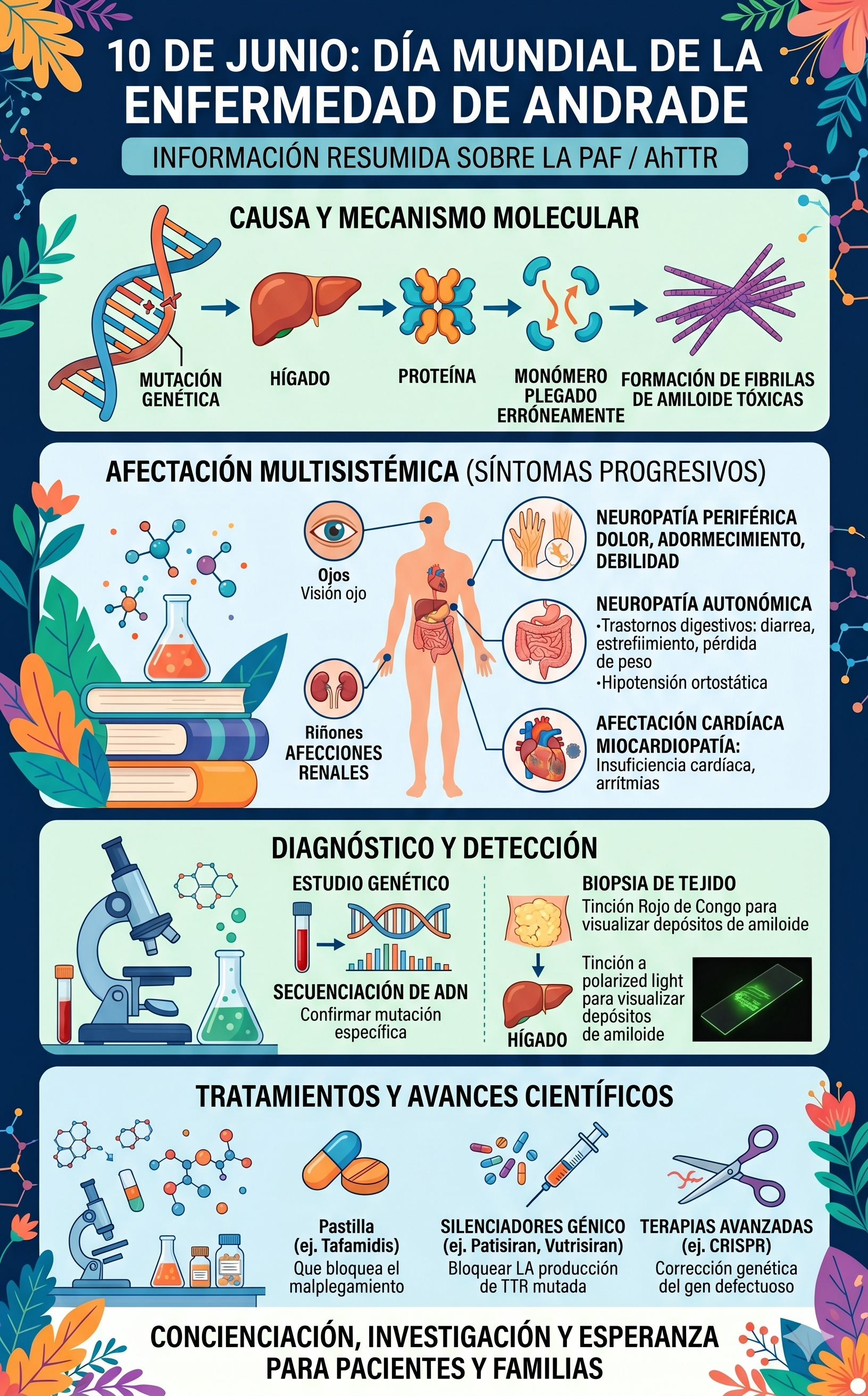
La enfermedad se desencadena por una mutación en el gen que codifica la proteína transtiretina (TTR), la cual se produce principalmente en el hígado y se encarga de transportar la vitamina A y la hormona tiroidea en la sangre.
El proceso de daño sigue los siguientes pasos secuenciales:
- Mutación genética: Provoca que la proteína TTR (que normalmente es una estructura estable de cuatro partes o tetrámero) se vuelva inestable.
- Descomposición: El tetrámero se rompe y se descompone en monómeros.
- Malplegamiento: Estos monómeros se pliegan de forma errónea debido a su defecto estructural.
- Agregación: Los monómeros defectuosos se agrupan formando agregados amorfos.
- Depósito de amiloide: Estos agregados se convierten en fibrillas de amiloide: fibras rígidas, insolubles y tóxicas que se depositan en los espacios extracelulares de los tejidos, destruyendo los órganos y nervios circundantes.
A nivel global existen más de 140 mutaciones descritas del gen TTR, pero la más común en España y Portugal es la denominada Val30Met (un cambio del aminoácido valina por metionina en la posición 30 de la proteína).
2. Síntomas y Progresión: Una Afectación Multisistémica
Los depósitos de amiloide dañan de manera progresiva los nervios periféricos (sensibilidad y movimiento) y los autonómicos (funciones involuntarias), además de órganos vitales. La afectación suele avanzar de forma ascendente:
- Extremidades inferiores (Neuropatía periférica): El debut suele consistir en hormigueos (parestesias), entumecimiento, pinchazos o una sensación constante de frío en los pies. Poco a poco se pierde la sensibilidad térmica y al dolor (lo que puede provocar quemaduras o heridas inadvertidas), seguido de debilidad muscular que dificulta la marcha.
- Aparato digestivo (Neuropatía autonómica): Provoca disfunciones severas como episodios de diarrea intensa intercalados con estreñimiento, náuseas, vómitos y una pérdida de peso extrema por malabsorción.
- Sistema cardiovascular: El amiloide se infiltra en las paredes del corazón, volviéndolo rígido (miocardiopatía restrictiva) y desencadenando arritmias, palpitaciones, bloqueos cardíacos o insuficiencia cardíaca.
- Otras manifestaciones: Pérdida de visión (por opacidad del humor vítreo o glaucoma), disfunción sexual, hipotensión ortostática (mareos graves o desmayos al ponerse de pie) y afectación renal.
3. Distribución Geográfica y el Impacto en España
A pesar de ser una enfermedad rara, su distribución es muy desigual debido al denominado "efecto fundador" genético. Los focos históricos más grandes del mundo se localizan en Portugal (Póvoa de Varzim), Suecia (Piteå) y Japón.
Sin embargo, España es una de las regiones con mayor prevalencia del planeta, destacando dos focos principales:
- Mallorca: Ocupa el quinto lugar a nivel mundial en volumen de casos detectados.
- Valverde del Camino (Huelva): Registra una de las densidades de población afectada más altas del mundo debido a la herencia familiar concentrada en la comarca.
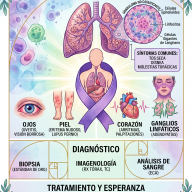
4. Diagnóstico: La Carrera contra el Reloj
Debido a que sus síntomas iniciales se confunden con los de otras neuropatías comunes (como la diabética), el diagnóstico solía demorarse entre 3 y 5 años. Hoy en día, la detección precoz es clave y se apoya en:
- Test genético: Un análisis de sangre permite secuenciar el gen TTR y confirmar la presencia de la mutación, incluso antes de que aparezcan los síntomas en familiares de pacientes.
- Biopsia de tejido: Se extrae una pequeña muestra de grasa subcutánea abdominal o de glándulas salivales menores. Al teñirse con Rojo de Congo y observarse bajo luz polarizada, los depósitos de amiloide muestran un brillo verde manzana inconfundible.
5. Panorama General de Tratamientos
El panorama terapéutico ha cambiado de forma radical. Históricamente, la única opción era el trasplante de hígado (para sustituir el órgano que fabrica la proteína mutada). Hoy en día, la farmacología ofrece alternativas avanzadas:
| Estrategia Terapéutica | Mecanismo de Acción | Ejemplos de Fármacos |
| Estabilizadores de la TTR | Se unen a la proteína normal e impiden que se rompa, evitando que forme los depósitos de amiloide. Eficaz en fases iniciales. | Tafamidis |
| Silenciadores Génicos (ARNi / Antisenso) | Bloquean el ARN mensajero en el hígado. Frena de raíz la fabricación de la proteína TTR (tanto la mutada como la normal). | Patisiran, Inotersen, Vutrisiran |
| Edición Genética (CRISPR) | Terapias diseñadas para "apagar" de forma permanente el gen defectuoso mediante una única infusión. | NTLA-2001 (en ensayos clínicos) |
Nota sobre las limitaciones actuales: Aunque las terapias de silenciamiento génico frenan drásticamente la progresión neurológica y cardíaca, la proteína amiloide residual sigue siendo difícil de combatir en áreas aisladas como el ojo o el sistema nervioso central, áreas donde la investigación médica actual sigue buscando soluciones combinadas.
6. Mecanismo Detallado del ARN de Interferencia (ARNi)
El ARN de interferencia (ARNi) es un mecanismo celular natural que los científicos han replicado en el laboratorio para "apagar" de forma selectiva la producción de proteínas específicas. En lugar de reparar el ADN o limpiar los depósitos ya formados, estos fármacos actúan en el paso intermedio: destruyen el ARN mensajero (ARNm), que es el plano de instrucciones que la célula usa para fabricar la proteína TTR.
El fármaco opera mediante el siguiente proceso celular:
- Direccionamiento al hígado: El fármaco está diseñado químicamente para viajar directamente a los hepatocitos (células del hígado), que es donde se produce el 99% de la TTR.
- Integración en el complejo RISC: Una vez dentro de la célula, la pequeña molécula de ARN bicatenario del fármaco se introduce en una maquinaria de proteínas propia de nuestro cuerpo llamada RISC (Complejo de Silenciamiento Inducido por ARN).
- Corte de precisión: El complejo RISC utiliza una de las cadenas del fármaco como guía molecular ("plantilla de reconocimiento") para buscar, emparejarse y "cortar" de forma específica el ARNm de la TTR.
- Destrucción del mensaje: Al fragmentarse el ARNm, los ribosomas celulares ya no pueden leer las instrucciones. Esto reduce los niveles de la proteína TTR en sangre entre un 80% y un 88%, cortando por completo el suministro de material para nuevos depósitos de amiloide.
7. Comparativa Técnica: Patisiran vs. Vutrisiran
Aunque ambos utilizan la tecnología de ARNi para silenciar el mismo gen, la forma en que se transportan dentro del cuerpo y su comodidad de uso presentan diferencias logísticas críticas:
| Característica | Patisiran (Onpattro) | Vutrisiran (Amvuttra) |
| Vehículo de entrega | Nanopartículas lipídicas (LNP): Envuelven el ARN en microesferas de grasa para protegerlo en la sangre hasta llegar al hígado. | Conjugado GalNAc: El ARN va unido directamente a un azúcar (GalNAc) que encaja a la perfección en los receptores específicos de las células del hígado. |
| Vía de administración | Infusión intravenosa (IV). | Inyección subcutánea (SC). |
| Frecuencia de dosis | Cada 3 semanas. | Cada 3 meses (cuatro veces al año). |
| Premedicación | Requiere corticoides, paracetamol y antihistamínicos previos para evitar reacciones a la infusión. | No requiere ningún tipo de premedicación. |
8. Resultados Clínicos y Eficacia Real
Los ensayos clínicos de Fase 3 han transformado por completo las expectativas de vida de los pacientes, demostrando que reducir drásticamente la TTR circulante no solo frena la enfermedad, sino que puede revertir parte del daño.
A. Impacto en la Polineuropatía (Daño neurológico)
- Patisiran (Ensayo APOLLO): Tras 18 meses de tratamiento, el 56% de los pacientes experimentó una reversión o mejora de sus síntomas de neuropatía (medido con la escala médica mNIS+7, que evalúa fuerza, reflejos y sensibilidad). El grupo placebo empeoró notablemente. También estabilizó la velocidad de la marcha y mejoró significativamente la calidad de vida.
- Vutrisiran (Ensayo HELIOS-A): Logró una reducción sostenida de la TTR en sangre del 88%. Demostró una eficacia equivalente a Patisiran, consiguiendo mejoras muy significativas en la marcha, el estado nutricional y la autonomía diaria de los pacientes frente a los datos históricos de control.
B. Impacto en la Miocardiopatía (Daño cardíaco)
- Vutrisiran (Ensayo HELIOS-B): Este estudio a gran escala evaluó el fármaco específicamente en pacientes con afectación cardíaca. Los resultados confirmaron que Vutrisiran redujo el riesgo de muerte por cualquier causa y de hospitalizaciones cardiovasculares en un 28% en la población general del estudio (y hasta un 33% en aquellos que lo usaban como terapia única en primera línea).
- Regresión del amiloide (Cambio estructural): Los análisis avanzados mediante resonancia magnética cardíaca revelaron que a los 3 años de tratamiento, el 22% de los pacientes mostró una reducción real de la acumulación de amiloide en el corazón (disminución del volumen extracelular), logrando recuperar elasticidad en el músculo cardíaco, algo que históricamente se creía imposible de lograr.
C. Seguridad y consideraciones
Ambos medicamentos muestran un perfil de seguridad muy alto. Los efectos secundarios más habituales son dolores articulares o musculares leves y reacciones locales transitorias en la zona de inyección. Debido a que se bloquea la proteína que transporta los nutrientes en el hígado, todos los pacientes bajo estos tratamientos deben tomar una suplementación diaria de vitamina A bajo estricto control médico para proteger su salud visual.
últimas noticias sobre Enfermedad de Andrade
Últimas noticias