FEDERACIÓN ESPAÑOLA DE ENFERMEDADES METABÓLICAS HEREDITARIAS
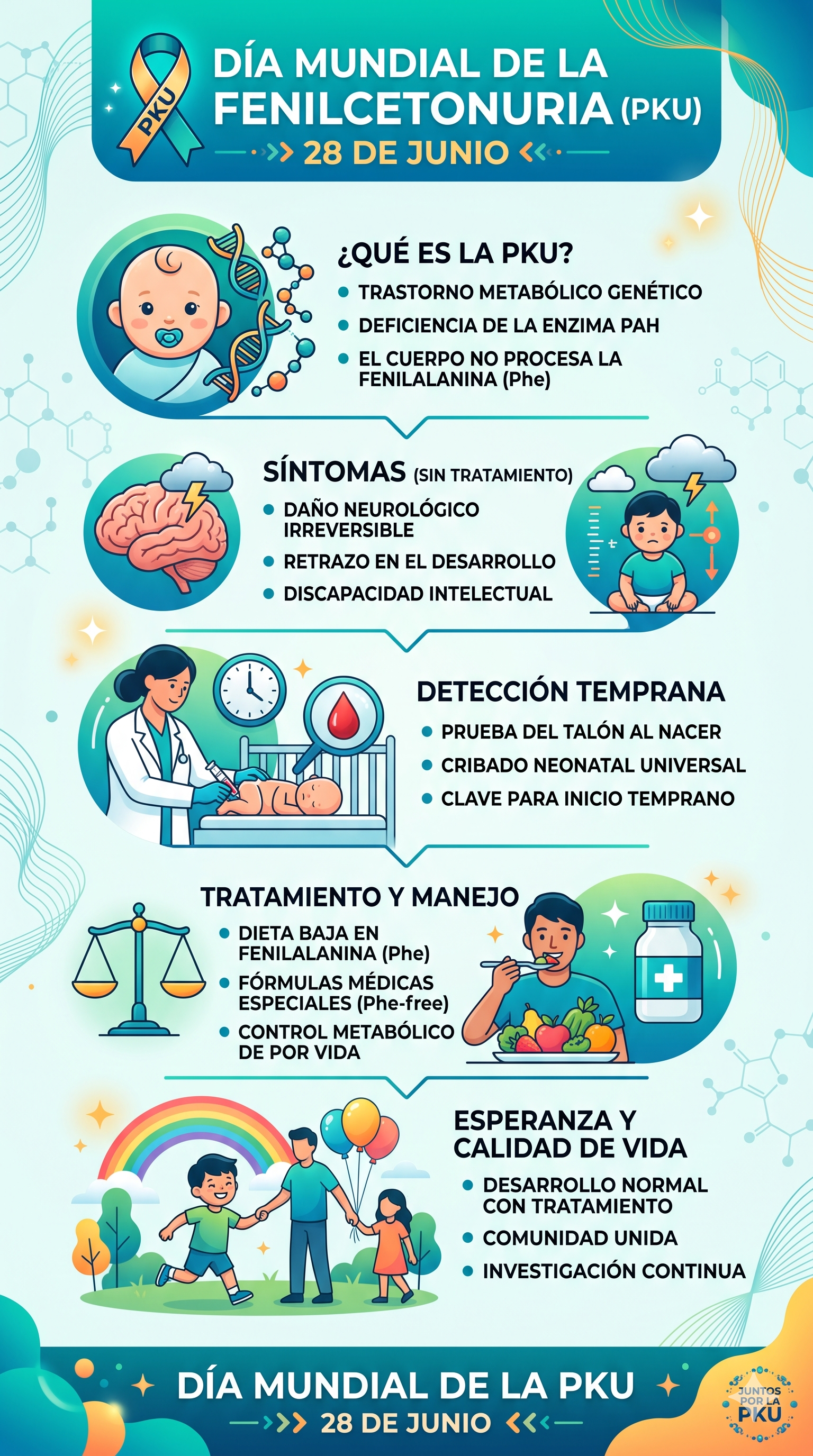
La fenilcetonuria (frecuentemente abreviada como PKU, del inglés Phenylketonuria) es uno de los errores innatos del metabolismo más estudiados e ilustrativos de la medicina contemporánea. Se trata de un trastorno genético con un patrón de herencia autosómico recesivo que altera la capacidad del organismo para procesar la fenilalanina, un aminoácido esencial que adquirimos estrictamente a través de la dieta.
A continuación, se presenta un desglose detallado, riguroso y sistemático de su fisiopatología, diagnóstico y abordaje terapéutico.
1. El Bloqueo Metabólico y su Genética
En un individuo sano, la fenilalanina ingerida que no se utiliza para la síntesis de proteínas se transforma en otro aminoácido, la tirosina. Este proceso ocurre principalmente en el hígado y es catalizado por la enzima fenilalanina hidroxilasa (phenylalanine hydroxylase). Para que esta reacción ocurra de forma óptima, la enzima requiere de un cofactor esencial: la tetrahidrobiopterina (BH4), además de oxígeno molecular.
La PKU está causada por mutaciones en el gen PAH, localizado en el brazo largo del cromosoma 12 ($12q23.2$). Al heredarse dos copias mutadas de este gen (una de cada progenitor), la actividad de la enzima es deficiente o nula.
Al interrumpirse esta vía, la fenilalanina se acumula de manera masiva en el torrente sanguíneo e induce una vía metabólica alternativa (transaminación), transformándose en metabolitos tóxicos conocidos como fenilcetonas (principalmente fenilpiruvato, fenillactato y fenilacetato), que terminan excretándose en altas concentraciones a través de la orina.
2. Fisiopatología del Daño Cerebral
El principal órgano diana de la toxicidad por fenilalanina es el sistema nervioso central. El mecanismo por el cual produce daño neurocognitivo irreversible si no se trata a tiempo se fundamenta en dos procesos críticos:
- Saturación del transportador LAT1: La fenilalanina comparte el mismo transportador en la barrera hematoencefálica (el transportador de aminoácidos neutros grandes o LAT1) con otros aminoácidos esenciales como el triptófano y la propia tirosina. Al haber concentraciones plasmáticas desproporcionadas de fenilalanina, esta satura por completo el transportador, impidiendo físicamente que el triptófano y la tirosina ingresen al tejido cerebral.
- Déficit de neurotransmisores e hipomielinización: Como consecuencia de la exclusión competitiva en la barrera, el cerebro sufre una carencia crítica de precursores para la síntesis de neurotransmisores clave. El déficit de triptófano reduce la producción de serotonina, mientras que la falta de tirosina deprime la síntesis de dopamina y noradrenalina. Adicionalmente, las altas concentraciones de fenilalanina interfieren de forma directa en la síntesis de mielina, provocando una desmielinización progresiva.
3. Manifestaciones Clínicas y Clasificación
Un recién nacido con fenilcetonuria suele ser completamente asintomático debido al aclaramiento metabólico que ejercía la placenta materna antes del nacimiento. Sin embargo, a medida que inicia la ingesta de leche (humana o de fórmula), la fenilalanina comienza a acumularse.
Si la enfermedad no se detecta ni se trata precozmente, los síntomas se manifiestan de manera gradual durante el primer año de vida:
- Discapacidad intelectual grave y global: Pérdida irreversible de puntos de cociente intelectual (CI) a una velocidad alarmante si no se interviene en las primeras semanas.
- Olor característico: Un olor corporal peculiar a humedad o "ratón", debido a la eliminación cutánea y urinaria del ácido fenilacético.
- Hipopigmentación: Los pacientes suelen presentar piel, ojos y cabello más claros que sus familiares biológicos. Esto ocurre porque la síntesis de melanina (el pigmento de la piel) depende directamente de la tirosina, la cual se encuentra severamente disminuida.
- Alteraciones neurológicas secundarias: Microcefalia, convulsiones, temblores y trastornos del comportamiento (hiperactividad o rasgos del espectro autista).
La gravedad del fenotipo clínico está directamente relacionada con el porcentaje de actividad enzimática residual, lo que permite clasificar la afección según los niveles plasmáticos de fenilalanina previos al tratamiento:
| Variedad Fenotípica | Concentración de Fenilalanina Plasmática | Actividad Enzimática Residual |
| PKU Clásica | Superior a 1200\text{ }\mu\text{mol/L}$ ($> 20\text{ mg/dL}$) | Menor al $1\%$ |
| PKU Moderada | Entre $600$ y $1200\text{ }\mu\text{mol/L}$ ($10 - 20\text{ mg/dL}$) | Del $1\%$ al $5\%$ |
| Hiperfenilalaninemia Leve | Entre $120$ y $600\text{ }\mu\text{mol/L}$ ($2 - 10\text{ mg/dL}$) | Superior al $5\%$ (suele no requerir restricción dietética estricta) |
4. Diagnóstico Precoz: El Cribado Neonatal
El pronóstico de la fenilcetonuria cambió radicalmente a mediados del siglo XX gracias a la introducción del cribado neonatal masivo (comúnmente denominado prueba del talón).
Coordenada temporal crítica: Para que la prueba sea plenamente fiable, la muestra de sangre capilar del talón del neonato debe extraerse idealmente entre las 48 y 72 horas de vida, asegurando que el lactante haya tenido una exposición proteica suficiente para detectar la elevación del aminoácido.
En los laboratorios bioquímicos modernos, el análisis se realiza mediante espectrometría de masas en tándem (MS/MS), una técnica de altísima sensibilidad y especificidad que cuantifica los niveles de fenilalanina y la relación fenilalanina/tirosina. Si se detecta una elevación sospechosa, se procede a una confirmación diagnóstica mediante la cuantificación formal en plasma y el descarte de anomalías en el metabolismo del cofactor BH4.
5. Abordaje Terapéutico y Nuevas Fronteras
El pilar fundamental del tratamiento de la PKU sigue siendo de carácter nutricional y debe mantenerse, de acuerdo con el consenso médico internacional actual, durante toda la vida.
Tratamiento Dietético
Consiste en una estricta restricción del aporte de fenilalanina a través de la dieta. Dado que este aminoácido se encuentra en todas las proteínas naturales, se deben eliminar por completo alimentos de alto valor biológico como carnes, pescados, huevos, lácteos, legumbres y frutos secos, así como productos que contengan el edulcorante artificial aspartamo (un péptido que libera fenilalanina al metabolizarse).
Para evitar la desnutrición proteica y el déficit de otros aminoácidos, los pacientes deben consumir diariamente fórmulas médicas especiales exentas de fenilalanina, pero suplementadas con tirosina, vitaminas, minerales y oligoelementos.
Alternativas Farmacológicas Modernas
El tratamiento ha evolucionado más allá de la dieta en subgrupos específicos de pacientes:
- Sapropterina (BH4 sintética): Actúa como una chaperona molecular. En pacientes que conservan cierta actividad enzimática residual (típicamente PKU moderada o leve), la administración de sapropterina estabiliza la enzima PAH mutada, potenciando su función y permitiendo una mayor tolerancia a las proteínas naturales.
- Pegvaliasa: Es una terapia de sustitución enzimática indicada para pacientes adultos que no logran un control metabólico óptimo con la dieta. Utiliza la enzima fenilalanina amonio liasa (derivada de bacterias y pegilada para reducir la respuesta inmunológica), la cual degrada la fenilalanina directamente en la sangre, independientemente de la funcionalidad de la PAH hepática o de la presencia de BH4.
El éxito clínico radica en iniciar estas intervenciones terapéuticas antes de que el lactante cumpla los primeros 10 a 14 días de vida. Si se mantiene un control metabólico riguroso de los niveles plasmáticos de fenilalanina dentro de los rangos de seguridad recomendados (generalmente entre $120$ y $360\text{ }\mu\text{mol/L}$), las personas con fenilcetonuria alcanzan un desarrollo neurológico, cognitivo y de esperanza de vida completamente normal.
últimas noticias sobre Fenilcetonuria
Últimas noticias





