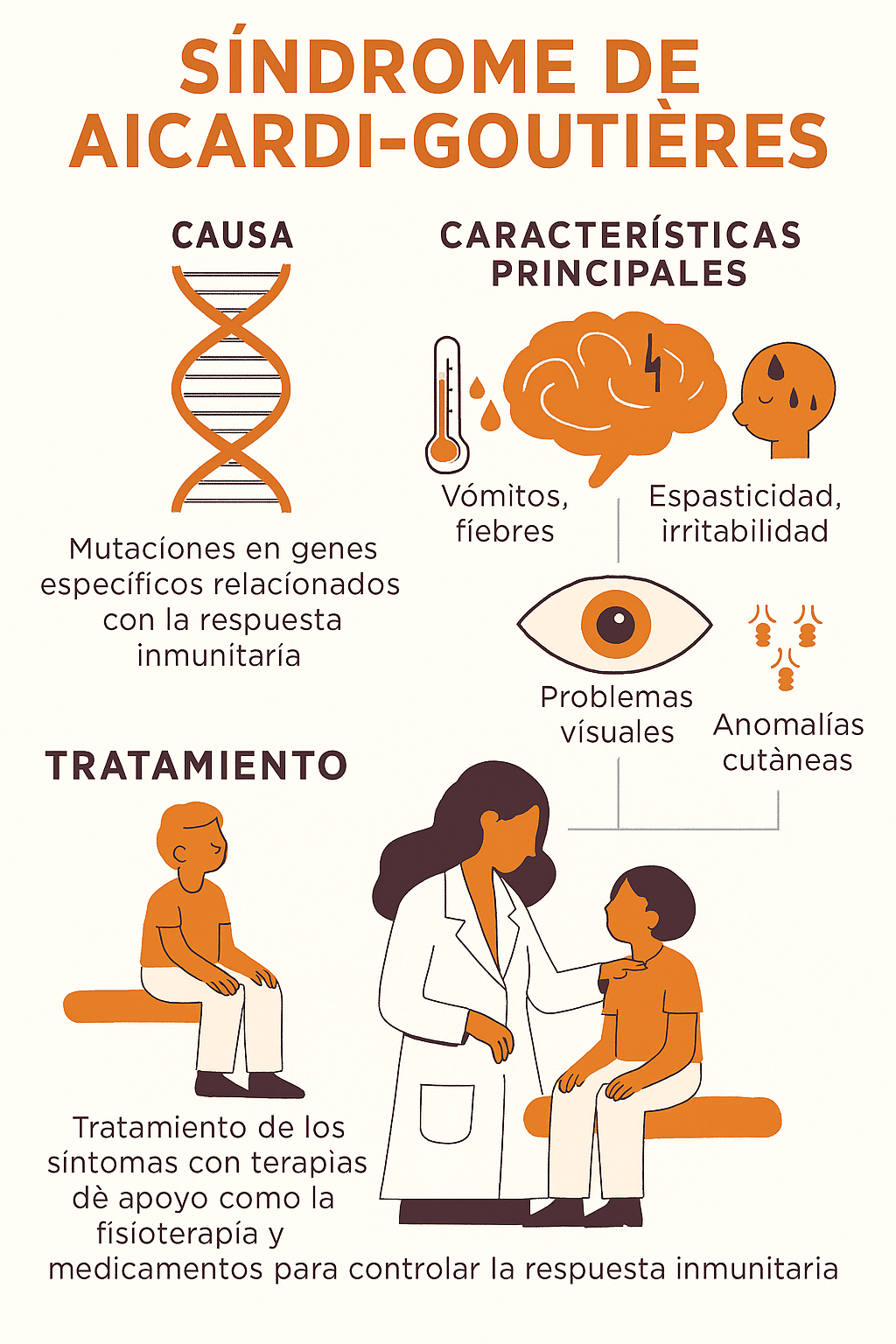
El Síndrome de Aicardi-Goutières (SAG) es una encefalopatía hereditaria rara que afecta principalmente al cerebro y al sistema inmunitario. Se clasifica como una leucodistrofia y es un ejemplo de enfermedad autoinflamatoria de origen genético.
Es crucial no confundirlo con el Síndrome de Aicardi (que se caracteriza por agenesia del cuerpo calloso, lagunas coriorretinianas y espasmos infantiles, afectando casi exclusivamente a mujeres).
A continuación, se detallan sus aspectos más importantes:
🧬 Causas y Mecanismo
El SAG es un trastorno genético con herencia principalmente autosómica recesiva (aunque algunos genes pueden tener patrones de herencia autosómico dominante).
-
Genes Implicados: Las mutaciones pueden ocurrir en varios genes que codifican proteínas relacionadas con el procesamiento de ácidos nucleicos (ADN y ARN), como:
-
TREX1
-
ADAR1
-
RNASEH2A, RNASEH2B, RNASEH2C
-
IFIH1
-
SAMHD1
-
-
Mecanismo Clave (Autoinflamación): Las mutaciones en estos genes provocan que el cuerpo no pueda deshacerse o procesar correctamente el ADN o ARN propios (endógenos). Esta acumulación de ácidos nucleicos mal procesados es interpretada por el sistema inmunitario como una infección viral.
-
Firma de Interferón Tipo I: La respuesta inmunitaria se caracteriza por una producción excesiva y crónica de Interferón tipo I (IFN-I) en el líquido cefalorraquídeo (LCR) y la sangre. Esta señalización persistente de IFN-I es la que causa el daño neurológico y sistémico, simulando una infección congénita viral (por eso se le llamó en el pasado "pseudotoxoplasmosis").
📉 Manifestaciones Clínicas
El SAG puede presentarse de dos formas principales:
1. Forma de Inicio Temprano (Más Común y Severa)
Se manifiesta en las primeras semanas o meses de vida con una encefalopatía subaguda.
-
Periodo Inicial: El bebé puede nacer aparentemente normal, pero pronto desarrolla:
-
Irritabilidad extrema y llanto inconsolable.
-
Fiebre sin causa infecciosa (fiebre estéril).
-
Pérdida de las habilidades psicomotoras adquiridas (regresión).
-
Dificultades para alimentarse.
-
-
Secuelas Neuromusculares: El deterioro conduce a una discapacidad intelectual y física grave, con:
-
Microcefalia (cabeza más pequeña de lo normal).
-
Espasticidad muscular y distonía.
-
Hipotonía troncal.
-
Convulsiones.
-
2. Forma de Inicio Tardío o Leve
El diagnóstico puede ser más tardío y los síntomas menos graves, a veces limitados a:
-
Trastornos neurológicos menos severos.
-
Sabañones (lesiones cutáneas de inflamación y enrojecimiento en dedos, pies u orejas, empeoran con el frío).
-
Afectación sistémica (tiroides, corazón, pulmones) sin gran encefalopatía.
Hallazgos Diagnósticos Clásicos
La tríada clásica que se busca en las pruebas es:
-
Calcificación de los ganglios basales y de la sustancia blanca (observable en la Tomografía Computarizada - TC).
-
Leucodistrofia (anomalía de la sustancia blanca del cerebro en la Resonancia Magnética - RM).
-
Linfocitosis en el LCR (aumento de glóbulos blancos en el líquido cefalorraquídeo, sin evidencia de infección bacteriana o viral).
🩹 Tratamiento
Actualmente, no existe una cura para el Síndrome de Aicardi-Goutières. El tratamiento es principalmente de soporte y sintomático:
-
Manejo Neurológico: Fármacos para el control de las convulsiones y la espasticidad.
-
Rehabilitación: Fisioterapia, terapia ocupacional y logopedia para maximizar la función y la calidad de vida.
-
Manejo Inmunológico: La investigación actual se centra en terapias que puedan bloquear la excesiva señalización de Interferón tipo I para reducir el daño inflamatorio.